

Roadmap: Unlocking machine learning for drug discovery

Advances in biology and medicine over the last decade have given us some of the most transformative medicines to date, including the first FDA-approved gene therapy, Luxterna, to treat retinal dystrophy, the first engineered cell therapy, Kymriah, to treat lymphoblastic leukemia, and multiple vaccines to treat the worldwide COVID-19 pandemic in less than a year since the virus was first discovered.
In 2020, the industry set a new NASDAQ biotech record by raising nearly $16 billion across more than 80 IPOs. By the second month of 2021, biotech IPOs had already raised nearly $3 billion cumulatively.
Despite these scientific breakthroughs and voracious investment appetite, the $1 trillion global pharmaceutical industry has been experiencing a productivity slide dating back to the 1950s. In just the last ten years, large cap biopharma companies have seen returns on investment in drug discovery decline from 10.1% to 1.8% and forecasted peak sales per drug cut in half. Pharmaceutical giants now often prefer deploying their cash reserves to partner with or acquire biotechnology startups versus investing heavily into internal R&D efforts; a third of forecasted sales are derived from acquisitions and a quarter from co-developed drugs. Many large cap biopharma companies are also facing the looming patent expirations on historical blockbuster drugs, leaving them actively searching for biotech startups and drug discovery methods to diversify their drug pipelines.
Over the last decade, Big Pharma has been betting on machine learning to usher in a new era of faster, cheaper, more-efficient, and advanced drug development. Despite some pharma industry concern that hype has exceeded reality in AI/ML drug discovery to date, we are now seeing the fruits of the pioneering work early evangelists have poured into the field. Most pharmaceutical giants have announced a partnership with a machine learning company, many firms are building machine learning R&D teams in-house, and the first drugs discovered via machine learning have now reached early-stage clinical trials.
We’ve previously written about how software is making the clinical trials process more efficient and remain interested in companies across the pharma IT value chain. Here, we share our perspective and roadmap on the entrepreneurial opportunities for machine learning in preclinical drug discovery engines.
The steadily rising cost of drug discovery
Moore’s Law, coined in 1965 by Intel co-founder Gordon Moore, predicted that computing power would double every 18 months. In drug development, we’ve witnessed the inverse. Pharmaceutical companies are spending increasingly more to develop fewer drugs. Eroom’s Law (Moore spelled backwards) states that the number of new drugs approved per billion U.S. dollars spent on R&D has halved roughly every nine years since 1950, an 80-fold drop in inflation-adjusted terms. Although estimates are hotly debated, today, of the five percent of drug candidates that make it to market, the development process costs roughly $2.5 billion.
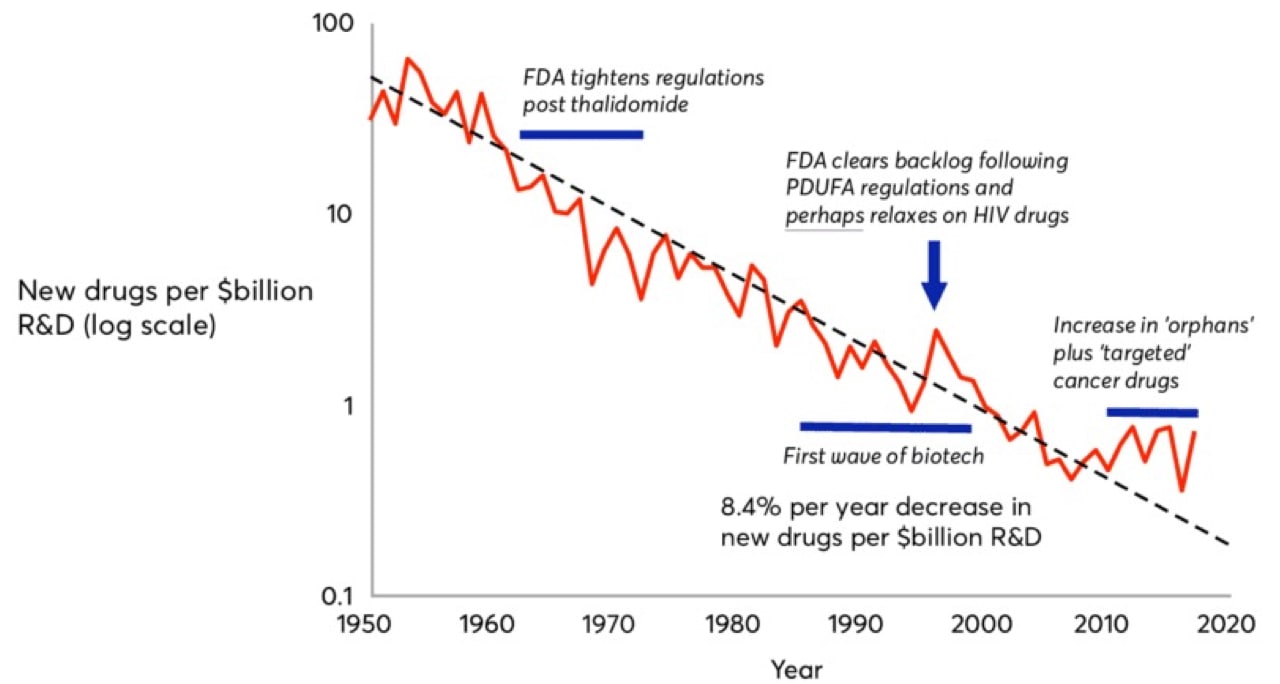
That $2.5 billion is composed not only of the direct R&D costs for the commercialized drug, but also the aggregate costs of all the failed drug candidates a drug company endured over the journey and the opportunity cost of investing their liquid assets elsewhere.
Why is drug discovery so expensive?
It’s in part inherent to the nature of biology. We still don’t understand biology well enough to build robust, bottom-up models of what proteins to target or how drugging a target of interest will affect an individual cell, much less an organ system or the entire human body. Most drug targets are parts of complex cellular networks leading to unpredictable changes (i.e. drug side effects). Biological systems also show a high degree of redundancy, which could blunt the effects of even the most specifically-targeted therapeutics.
It’s also inherent to the nature of the market. Commercially-viable drugs have to be significantly better than currently available treatments, so as more drugs are approved, building a new drug for a disease with prior treatments becomes increasingly more difficult. This explains why many pharma companies have focused heavily on rare diseases and tumors with unique mutations (known as precision oncology), where a new drug may be the first and only treatment available.
The last two decades have also experienced bias towards brute force approaches to drug discovery. The industry has shifted from iterative medicinal chemistry coupled with phenotypic assays to the serial filtering of a static compound library against a given target. As a result, we’ve overestimated the ability of high-throughput screening of large chemical libraries to a specific target to tell researchers that a drug candidate will be safe and effective in human clinical trials.
In fact, more first-in-class small-molecule drugs in the first half of the 2000s were discovered using old-school phenotypic assays versus today’s drug-target affinity assays, even though the latter has been the new industry standard for the past two decades. Phenotypic screens, although slow, capture and abstract away the complex network of unpredictable effects from drugging a target, which is impossible to identify in a lone drug-target binding affinity assay. Capturing, representing, and perturbing that complex biological network in silico is the holy grail of machine learning for drug discovery.
Deconvoluting the drug discovery process
Drug discovery, the process of identifying novel therapeutics to treat disease, is the first component of the biopharmaceutical value chain. We’ve covered ways how software can make the latter components of the biopharmaceutical value chain, including clinical trials, more efficient elsewhere. The drug discovery process may differ based on whether a drug candidate is a small molecule or a biologic (e.g. proteins, antibodies, cell therapies, gene therapies). The majority of ML approaches to drug discovery to date have focused on small molecule development, although recent companies have emerged to tackle biologics.
The discovery process begins with studying the biology of a cellular target, i.e. something a drug can ‘hit’. Once the biology is well characterized, the process continues with high throughput screening of thousands of existing chemical compounds to find those that ‘hit’ the target of interest, known as leads. Next, scientists make iterative chemical changes to lead molecules to maximize properties of interest, such as binding affinity to the target, and test the distribution, clearance, and toxicity of the drug in a petri dish and animal models.
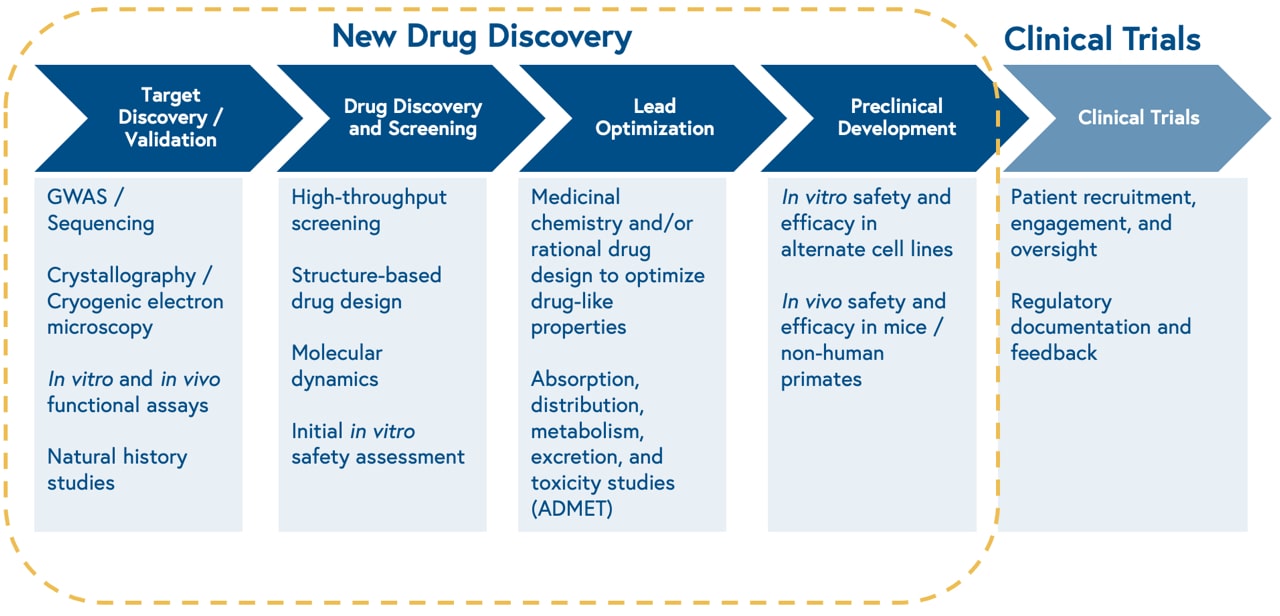
This approach is slow, limited by the size of physical chemical compound libraries, and prone to high attrition rates. Consequently, only one out of 10,000 molecules screened for a given target will make it through the drug development process.
Why now?
The pharmaceutical industry is riding tailwinds from scientific revolutions in both biomedicine and computer science. There are three drivers highlighting why now is the key time to focus on the potential of machine learning in drug discovery.
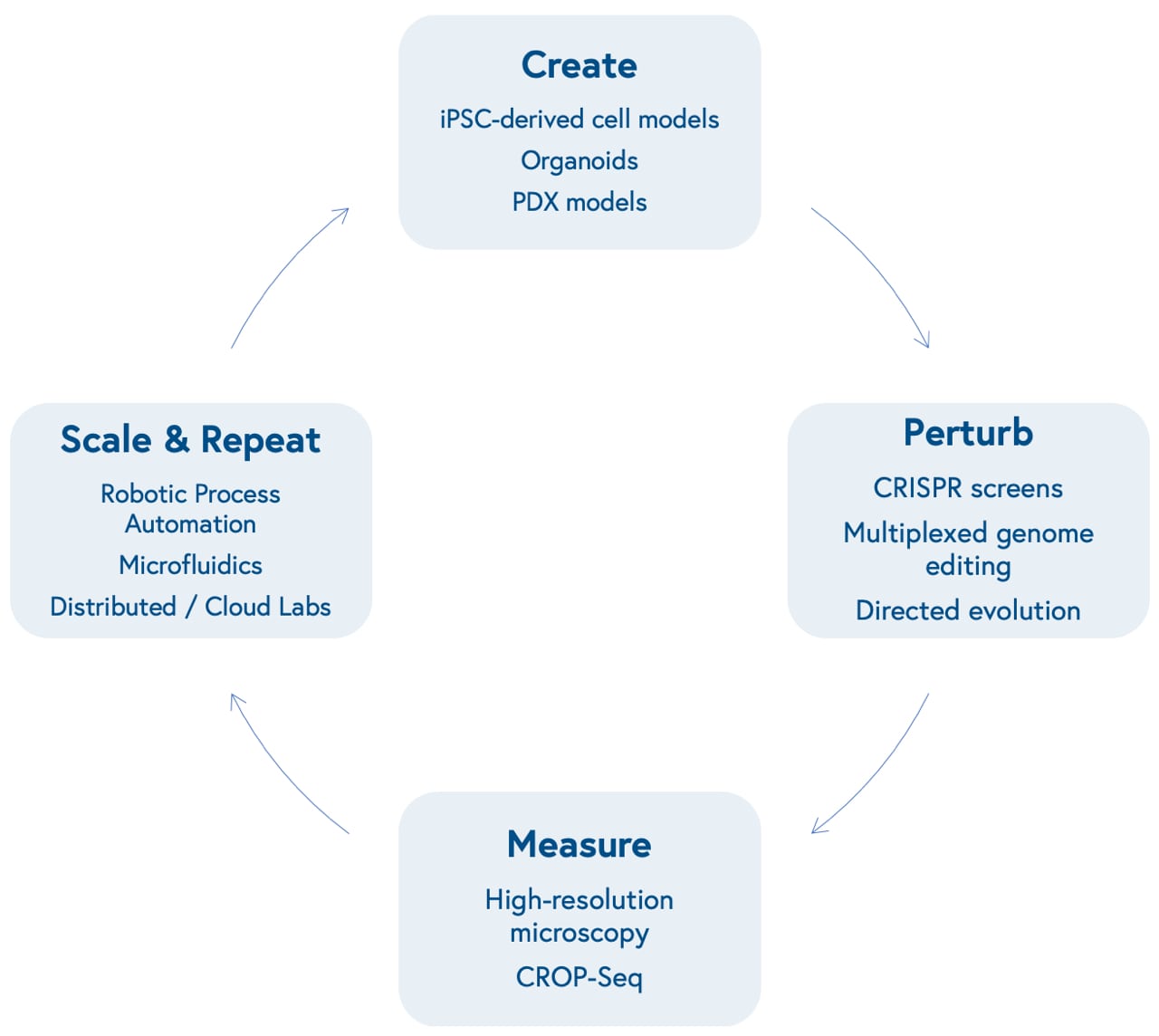
1. New tools have improved our ability to create, manipulate, and measure biological systems at scale.
Advances in molecular biology and bioengineering over the past decade have improved our ability to represent human biological systems in smaller sizes and test therapeutic hypotheses at scale. This begins with our ability to create models that more accurately reflect unique biological dynamics and variation, including induced pluripotent stem cell (iPSCs)-based models of different cell types, organoids that reflect three-dimensional tissue dynamics, and patient-derived xenografts (PDX) to mimic human disease in mouse models.
Next is our ability to perturb these models in a genome or protein-targeted fashion. The development of multiplexed CRISPR/Cas9-based gene editing gave researchers the power to quickly insert, delete, modify, or replace multiple genes in living organisms.
CRISPR screens allow scientists to activate or knock out the function of one or many specific genes at once in high-throughput and identify genetic dependencies unique to certain cell types (known as synthetic lethality). Directed evolution of proteins allows scientists to mutate structures to yield advantageous characteristics, such as tissue or biomarker-specific targeting and evasion of the immune system.
Subsequently is our ability to measure those perturbations at incredibly high-resolution. This can be accomplished through advances in microscopic imaging, including the ability to visualize live-cell dynamics, or more granular genetic sequencing, including the ability to measure changes in gene expression level at the single-cell level (single-cell RNA sequencing) after perturbations with CRISPR screens (CRISPR droplet sequencing, or CROP-Seq).
Finally is our capacity to scale and repeat this cyclical process. Robotic automation and microfluidics within in-house labs or via virtual, distributed labs, including contract research organizations and virtual labs like Strateos, Emerald, and Culture Biosciences allow biotech entrepreneurs to outsource and scale otherwise manual, tedious, and repetitive biological experiments, making it easier and cheaper to produce proprietary biological data. These data may offer new insights into biological systems, providing researchers with the knowledge to create new models that reflect varying diseases and repeat the loop.
2. Machine learning models are delivering better representations of biological systems
In the realm of software, the cloud and machine learning infrastructure that has been a cornerstone in SaaS and redefined dozens of industries is making its way to biopharma. Newer model architectures, such as gated graph sequence neural networks and quantum machine learning, are making progress towards better characterizing the spatial, electrical, and energetic states of drug candidates, their biological targets, and the high-dimensional biological systems they interact within.
Fueling those representations are larger and more granular biomedical, chemical, and clinical data collection and distribution efforts on the part of the government, hospitals, universities, and biotech firms. Most notable of these data collection efforts is the National Institute of Health’s ‘All of Us’ research program, launched in 2018, which is actively collecting longitudinal health and genetic data from 1 million volunteers to help scientists make progress in precision medicine. A notable challenge with publicly aggregated data sources (e.g. PubChem, ChEMBL, TCGA, ImmPort) is that the raw datasets are often incomplete, noisy, unstructured, and poorly understood, contributing to well-known ‘garbage in, garbage out’ concerns. The last decade, however, has benefited from a surge in open-source bioinformatics tools to clean datasets for functional use as inputs into statistical machine learning models. Made possible by the advances in biology mentioned earlier, both large biopharma firms and young startups are building end-to-end approaches to training machine learning algorithms, generating proprietary, high-quality data at the lab bench to feed into algorithms to build better predictive models.
3. Bilingual talent is applying computing power to ask the right questions of complex biological systems
A new breed of entrepreneurs, including bilingual teams that closely pair engineers with biochemists, and scientists trained in both biology and computer science, are paving the way for machine learning to ask the most compelling questions of the biological data we have on hand.
The partnership between scientists, machine learning engineers, and clinicians not only asks what problems are worth solving, but also which problems can we solve and what data do we need?
For all its promise, machine learning in biomedicine will depend on human talent to set research directions, generate novel biological questions and hypotheses, produce and filter data, and validate results. We’re likely to see hybrid models with bespoke assays run by scientists and standard experiments run by machines and software.
Opportunities for machine learning in drug discovery
Machine learning applies algorithms to learn from data and then either characterizes or makes predictions about new data sets. Three factors influence the potential of machine learning to make useful prediction in drug discovery: 1) the specific features of a molecule, target, or biological system available as input data, 2) the quality and quantity of data available, and 3) selection of the right model architecture for the task at hand.
Machine learning algorithms can take a variety of biological features as inputs, including genetic sequencing data, small molecule structure libraries, biochemical assay data, microscopy images, or text from academic literature. These models can largely be broken up into either supervised or unsupervised learning. Supervised learning aims to predict future outputs of data, such as regression and classification, whereas unsupervised learning aims to identify novel hidden patterns or relationships within high-dimensional datasets and cluster similar data together. There are opportunities for both in the drug discovery value chain.
Drug Discovery Value Chain

1. High-throughput in silico analyses of multidimensional data could identify novel biological targets

Drug discovery relies on the development of drug candidates, including small molecules, peptides, antibodies (and more recently nucleic acids and cells) designed to alter disease states by modulating the activity of a molecular target of interest. Identifying a target of interest relies on a therapeutic hypothesis, i.e. that modulating a given target will lead to a change in the disease state.
Novel biological measurement tools, including high-dimensional microscopy and single-cell RNA sequencing are well-suited as multidimensional training data for unsupervised machine learning models to infer new, meaningful relationships between biological components, i.e. potential targets, and disease.
We have encountered two (not mutually exclusive) classes of companies applying machine learning methods in this space:
- Precision medicine (i.e. -omics-based) startups sifting through a combination of public and self-generated genomic, transcriptomic, and proteomic data to identify and validate novel targets (e.g. Insitro, Octant, Deep Genomics, Verge Genomics). Within this class are two additional approaches: Combination target discovery: identification of genomic and metabolomic correlates of response to standard-of-care treatments, such as immune checkpoint inhibitors (e.g. Ikena Oncology, Immunai). Synthetic lethality: identification of paired genomic targets that, when perturbed or mutated in tandem, lead to cell death (e.g. Tango Therapeutics, KSQ Therapeutics, Artios Pharma).
- Imaging-based startups applying convolutional neural network architectures to high-resolution digital microscopy to evaluate phenotypic cellular changes when perturbed with a drug candidate of interest. As mentioned earlier, this high-throughput phenotypic approach captures and abstracts the complex network of unpredictable effects (i.e. polypharmacology) that scientists may otherwise miss in modern drug-target affinity assays that evaluate one compound against one target at a time. (e.g. Recursion Pharmaceuticals, Eikon Therapeutics).
Furthermore, as modern biomedical science becomes increasingly rich in data, products like Watershed Informatics are commoditizing bioinformatics and machine learning tools to enable both academic biologists and pharmaceutical companies to quickly run initial target discovery analyses on raw sequencing data.
Finally, biomedical literature is the primary setting of our collective knowledge of associations between molecular targets and disease. Companies like nference are processing unstructured text with natural language processing (NLP) to identify relevant papers and associations between diseases, targets, and drugs among the trove of digitized literature. This approach has been particularly useful for identifying new applications of previously commercialized drugs or shelved drug candidates whose data have been described largely in free text.
2. Model architectures that represent biochemical structures, properties, and interactions with potential targets could identify more promising drug candidates

Typical small molecule drug discovery involves experimental, laboratory-automated, high-throughput screening of millions of diverse chemical compounds against a protein target of interest to identify ‘hits’. Hits are then biochemically modified to optimize target specificity, selectivity, and binding affinity.
Representing these three-dimensional structures and thermodynamic interactions between drugs and targets in silico is both mathematically and computationally challenging. Laboratory approaches to identify and visualize protein structures, including X-ray crystallography and cryogenic electron microscopy are expensive and difficult, and, even when a structural representation of a molecule is available, simulating protein movement/dynamics is computationally demanding.
In the 1980s, the biopharma industry developed early versions of statistical and biophysical modeling programs for molecular docking prediction. These were limited by both limited computing power and an incomplete understanding of how molecules interact. New unsupervised learning methods and novel model architectures to represent molecules, such as graph convolutional networks, can derive their own insights about which biochemical features matter to develop better predictions of protein structure and drug-target interactions.
The most notable recent computational development in structural biology came late last year, when DeepMind’s AlphaFold2 algorithm was able to reliably predict over 90% of a single protein structure based on the amino acid sequence alone. While this feat is not at the level of resolution needed for target discovery yet, it remains a promising advance for the future of protein structure prediction. More immediately, technologies like AlphaFold2 may be useful for designing protein-based therapeutics, including antibodies and peptides, where high-resolution prediction is less necessary.
Startups today are leveraging new model architectures, vast public and proprietary biochemical structure and assay data, and novel biological tools to identify more promising hits, i.e. machine learning can give us better starting molecules to move through the drug development process. We’ve categorized companies tackling this problem into three groups:
- Deep learning-guided simulations of molecular docking, i.e. the spatial interaction and binding affinity between a potential drug and the target protein. (e.g. Atomwise, BenevolentAI, Nimbus Therapeutics). Within this class are two additional approaches: Structural Allostery: Applying computational biophysics, genomics, and molecular dynamics for identification of novel, non-competitive (allosteric), binding sites on target molecules (e.g. Relay Therapeutics, Hotspot Therapeutics, Frontier Medicines). DNA-encoded Libraries (DELs): allow biopharma companies to inexpensively predict drug activity from up to 40 trillion molecular structures, stored in a single test tube mixture, by linking distinct DNA sequences as unique barcodes for each tested biochemical compound. Emerging biotech companies are using these data to train machine learning models to predict drug activity on more diverse compounds that weren’t included in the DELs (e.g. ZebiAI, Anagenex).
- Molecular de novo design via generative model architectures, including recurrent neural networks, variational autoencoders, and generative adversarial networks, have been used to create small molecules and protein therapeutics with optimal bioactivity, pharmacokinetics, and other desired properties (e.g. Insilico Medicine, Generate Biomedicines).
- Repurposing or identifying synergistic combinations of off-patent drugs, which by nature of commercial use have de-risked safety profiles, to modulate novel targets (e.g. Pharnext, BioXcel Therapeutics).
3. In silico prediction and optimization of pharmacodynamics / pharmacokinetics via better in vitro microphysiological systems could improve the success rate of drug candidates in clinical trials.

Lead optimization involves rational chemical modification and evaluation of lead compounds for desired properties, including pharmacodynamics (the effect of the drug on living organisms) and pharmacokinetics (how the living organism acts on the drug) both in silico and in vitro. Pharmacokinetics is better described by the acronym ADMET: absorption, distribution, metabolism, excretion, and toxicity (i.e. off-target effects). For each lead, this process involves synthesizing and testing a large number of similar molecules in silico and in vitro.
In silico prediction of undesirable off-target effects of leads and associated idiosyncratic toxicity could minimize the number of eventual failures of drug candidates in human clinical trials. This has long been a challenge that Big Pharma has gone after, and companies like Pfizer, Bayer, Sanofi, and Bristol-Myers Squibb have all published their in silico approaches to lead optimization and ADMET prediction. Improved in vitro microphysiological systems, such as three-dimensional organoids and organ-on-a-chip devices, offer a higher quality and more biologically-relevant data source for predictive models compared to traditional petri dish assays.
Startups in this space are both evaluating public assay data to predict optimal chemical modifications for lead candidates and building high quality in-house bioactivity and ADMET datasets themselves (e.g. Reverie Labs, Genesis Therapeutics).
4. Computational prediction of biomarkers correlated to drug response in vitro and in vivo may improve the success rate of drug candidates in human clinical trials.

Optimized compounds are finally tested in an in vitro model of disease, from cell lines to organoids, to determine how well the drug candidate is modulating the target of interest within a living system. Safety and efficacy of the drug candidate is then tested in varying animal and patient-derived xenograft (PDX) models, ranging from mice to non-human primates, depending on the target and disease of interest. However, this process is expensive, time-consuming, and only weakly correlated to how well the drug will fare in humans. Some startups were built to serve biopharma companies faster than traditional contract research organizations with automated collection of animal model data (e.g. Vium, acquired by Recursion).
To help improve the success rates of clinical trials, biotech companies will often employ unsupervised learning to identify translational biomarkers in these preclinical models that predict better response to a drug candidate. This has been particularly useful in the case of precision oncology and immunotherapy, in which genomic signatures have predicted drug sensitivity in a tissue-agnostic manner. Identifying predictive biomarkers can improve selectivity in clinical trial recruitment and stratify patients in later-stage trials (e.g. Scorpion Therapeutics, Volastra Therapeutics). Additionally, companies like PathAI and Paige are supporting biopharma with computational pathology as a service, identifying spatial biomarkers that correlate to drug response.
New frontiers
Developing in silico models that deliver realistic, meaningful representations of life remains a major challenge in biology and represents a large opportunity for scientists and engineers. Two emerging areas of interest for machine learning we’re interested in are in de novo protein engineering and quantum mechanics-informed simulations of molecular dynamics.
Protein engineering through machine-guided directed evolution enables scientists to optimize desired protein features through the creation of novel variants. These methods predict how protein sequences map to function without requiring prior knowledge of the underlying protein structure, molecular dynamics, or associated biological pathways.
A key trend in biopharma has been the growth of biologics, i.e. protein-based therapeutics such as antibodies and enzyme replacement therapy, due to lower attrition rates, strong safety profiles, and defensibility from competing biosimilars relative to small molecules, which more easily lose market share to generics.
Companies like Manifold Bio, LabGenius, Serotiny, and Nabla Bio are working on machine learning-based approaches to protein engineering. AbCellera and BigHat Biosciences are applying these same principles directly to the development of more selective and less immunogenic antibody therapeutics. Finally, Dyno Therapeutics has carved a niche in designing bespoke, machine intelligence-informed adenovirus-associated vectors (AAVs) to preferentially deliver gene therapies to specific organs and avoid provoking an immune response to the viral vector itself, which has troubled gene therapies of years past.
As computing power continues to scale in the cloud year over year, companies like Silicon Therapeutics (acquired by Roivant Sciences) and ProteinQure are developing quantum mechanics engines to explore interactions beyond drug-target binding, including loop dynamics and domain breathing, which may more accurately represent specific, complex conformations of targets and identify unique binding sites for drug candidates in silico.
The response to the COVID-19 pandemic by both software and biopharma is a testament to the resilience and ingenuity of both industries, and we believe there is no better time than now to see their core strengths converge to develop the next-generation of lifesaving therapeutics.
We are excited to continue partnering with and learning from the scientists and engineers working on novel computational approaches to design better medicines faster.
If you’re building something to accelerate the speed and productivity of drug discovery, we want to hear from you. (Email Andrew Hedin at ahedin@bvp.com and Nisarg Patel at npatel@bvp.com).


